
-
新 EU 의료기기 규정, 하반기 전면 시행에 미리 대비해야
- 통상·규제
- 독일
- 함부르크무역관 Sumin Moeller
- 2017-03-17
- 출처 : KOTRA
-
- 독일 의료기기시장 꾸준한 성장, 한국제품 수출도 증가세 -
- CE 규정 개정으로 기술문서, 라벨링 및 인증요건 등 강화, 우리 기업의 철저한 대비책 필요 –
□ 독일 의료기기 시장
ㅇ 2017년 4%대의 꾸준한 성장 예상- 세계 3위인 독일 의료기기 산업은 2015년 전년대비 11% 성장해 284억 유로 규모의 매출을 기록했으며, 지속적인 성장세를 이어갈 것으로 예상됨.
* 독일 의료기술협회(BVMed)의 2016년 10월 조사에 따르면 2016년도 해당 산업의 성장률은 4%에 달할 것으로 분석됨.
- 약 1만2500개 기업에 19만5000명이 관련 업계에 종사하고 있으며 독일 의료기술 기업의 대부분(전체의 94%)은 250명 이하의 직원을 보유한 중소기업들임.
- 혁신기술 집약적, 짧은 제품 수명주기 등과 같은 산업의 특성상 전체 매출의 1/3 정도가 창업 3년이 되지 않은 신생 기업에 의한 것이며, 평균적으로 매출의 9%를 R&D에 투자하고 있음.
- 2016년 독일 의료기술산업 총매출의 68%가 수출에 의한 것임.
독일 의료기술 산업 매출 추이(단위: 백만 유로)
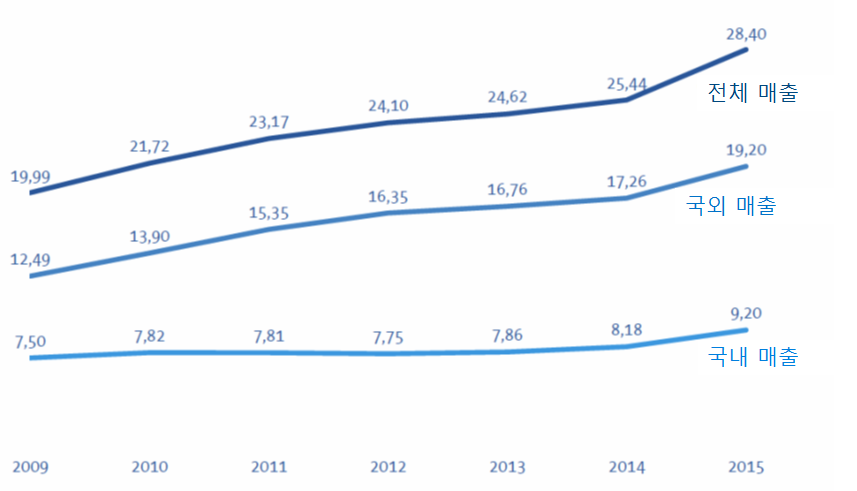
자료원: 독일의료기술협회
ㅇ 독일 시장은 한국의 제2위의 수입 시장인 동시에 제3위의 수출시장임.
- 2015년 기준 대독일 수출은 전년대비 14% 증가한 2억2700만 달러, 수입은 9% 감소한 4억4200만 달러로 총 교역액 6억7000만 달러를 기록
- 2016년에는 전년대비 정형외과용 기기(33%), 기타 호흡용 기기(48%), 엑스선 촬영기(10%)가 수출 호조를 보였으며, 특히 자기공명 촬영기(578%), 치과용 드릴엔진(1171%), 컴퓨터 단층촬영기(6808%)는 매우 큰 폭의 수출 증가율을 달성함.
* 이에 대독일 무역수지 적자는 25% 줄어든 2억1500만 달러를 기록
- 독일 의료기기 주요 수입국으로는 미국, 스위스, 중국, 일본, 네덜란드 프랑스 등이 있음.
독일 의료기기 산업 주요 국별 수입 동향
(단위: 천 달러, %)수입국
2014년
2015년
2016년
2015/16 증감률
미국
2,662,367
2,655,045
2,643,862
-0.4
스위스
1,962,376
1,918,117
1,993,308
3.9
중국
932,514
928,346
981,527
5.7
일본
953,559
800,770
853,006
6.5
네덜란드
749,905
672,035
692,759
3.1
프랑스
740,889
640,185
648,588
1.3
영국
659,264
640,396
574,339
-10.3
폴란드
414,314
387,928
405,520
4.5
벨기에
278,366
216,196
203,333
-5.9
한국
135,027
117,914
124,966
6.0
주: HS 코드 9018(진단기기, 기타 전기의학의 장치 및 검안기기를 포함하는 내·외·치과 또는 수의과용 기기),
9019(기계요법용 기기, 치료용 호흡기), 9020(기타 호흡기기), 9021(정형외과용 기기), 9022(엑스선 촬영기)의 합계
자료원: 글로벌 트레이드 아트라스□ CE 규정 개정 배경 및 진행 현황
ㅇ CE 규정 개정 배경
- 2012년 프랑스 PIP사 인공 유방 보형물에 공업용 실리콘으로 만든 발암성 유방 성형 보형물이 사용된 사건
- 이로 수십 명이 암에 걸리고, 이를 이식 받은 수십만 명이 검사를 받는 사태가 발생
- 이를 계기로 유럽 내 심사기관(Notified Body)의 품질과 역량개선을 위한 유럽 의료기기 CE 제도 실행 및 개선 강화의 필요성 대두
- 사건 직후 EU 집행위원회가 1990년대 제정된 법규의 개정안 초안을 내놓았음. 그러나 업계의 반대 로비, 소비자 단체들의 비판, EU 회원국 간 이견 등으로 표류
- 결국 의회가 중심이 돼 이해 당사자 및 EU 관련 기관들과 협의·조정을 거침. 합의안을 만드는데 4년의 시간이 걸려 2016년 7월 상임위 통과했으며 2017년 시행을 앞두고 있음.
ㅇ 기존 독일규정 전면 재편
- 새로 발효된 법규(Regulation, 독어 Verordnungen)는 EU에서 최상위 법규로 회원국 전체에 일괄적용할 예정이며, 기존 독일의 의료기기 지침(독어 Richtlinie)보다 상위법으로 기존 지침들은 대체될 예정
- 기존 독일에는 의료기기에 관한 아래의 3가지 지침 존재
▶ 일반 의료기기 지침 93/42/EWG(Medical Device Directive, MDD)
▶ 능동 이식형 의료기기 관련 지침 90/385/EWG(Active Implantable Medical Devices, AIMD)
▶ 체외 진단 의료기기 지침 98/79/EG(in-vitro Diagnostika, IVD)
- 일반 의료기기 지침(MDD)와 능동 이식형 의료기기 지침(AIMD)은 EU 의료기 규정(Medical Device Regulation, MDR)로 편입, 통합될 예정
- 체외진단 의료기기 지침(IVD)은 EU 체외진단 의료기기 규정(IVDR)으로 편입될 예정
- 이에 2017부터 독일 시장은 물론 유럽 시장에 판매되는 의료기기는 의료기기 규정(MDR), 체외진단 의료기기 규정(IVDR)을 따라야 함.
독일 의료기기 규정 개편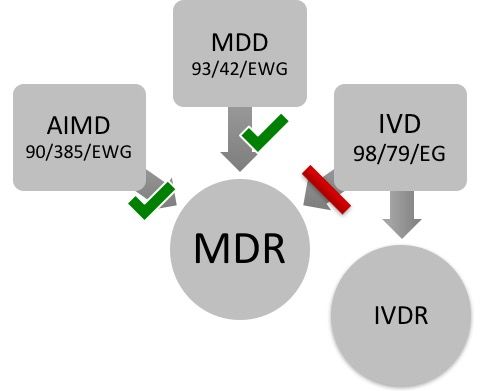
자료원: Johner Institut
ㅇ 진행현황 및 예상 진행 상황
- 2017년 2월 22일 MDR의 최종적 틀을 EU 의회 제출
- 3월 7일 EU 의회 내 첫 번째 독회
- 4월 중 EU 의회 두 번째 독회 예정
- 2017년 하반기 법규 공시 및 20일 이내 시행(2017년 7월로 예상)
□ CE 규정 개정 1: 의료기기 규정(MDR)
ㅇ MDR 구성
- 유예기간 3년(2017~2019년)을 가지며 10장, 17부록으로 구성
* 10장(1. 사용분야 및 정의, 2. 제조업체, 유통업체 및 회원국에 대한 요구사항: 적합성평가 절차, 라벨 시판 후 임상추적, 사후 시장 감시, 3. 제품 추척성, 4. 승인기관에 대한 요구사항, 5. 분류 및 적합성 평가, 6. 임상 평가 및 임상 시험, 7. 시장 감시 및 규제 리포팅, 8. “Medical Coordination Group“과 다른 전문가 집단, 회원국 간의 협력, 9. 기밀성, 데이터 보호, 처벌, 10. 유예 기간, 그 외)
* 17부록(1. 일반 안전 및 성능 규제, 2. 기술 문서, 3. 시판 후 감시에 대한 기술 서류, 4. EU 선언의 적합성, 5. CE 마크의 적합성, 6. 기기 등록을 및 고유식별코드를 위한 정보, 7. 인증기관의 요구사항, 8. 분류 기준, 9. 품질 관리 및 기술 서류 평가에 대한 적합성 평가, 10. 타입 검사에 따른 적합성 평가, 11. 제품 적합성 검증에 따른 적합성 평가, 12. 주문 제작 기기에 대한 절차, 13. 인증기관에서 발급한 인증서, 14. 임상 평가 및 PMCF, 15. 임상 연구, 16. 의료 목적이 없는 제품 그룹 목록, 17. 관련 테이블)
ㅇ 주요 변경 내용
주요 변경 사항
세부 내용
기술 문서 강화
부록 2의 기술문서 부분이 훨씬 세분화 됨은 물론, 지속적인 업데이트가 요구됨
식별, 추적성 향상
모든 의료기기 제품은 고유식별코드(Unique Device identification, UDI)가 적용돼야 함
라벨링
새로운 제품 라벨링 규제 적용
유럽 의료기기 데이터베이스 (EUDAMED)
유럽 의료기기 데이터베이스(EUDAMED) 확장으로 등록 준비가 요구됨. 현재까지 이 데이터베이스는 국가 기관에서만 접근 가능했으나 제조사, 인증기관, 일반 대중에 공개될 예정
새로운 시험과정 도입
고위험 의료기기에 대한 심사를 위한 전문가 자문기구(Medical Device Coordination Group, MDCG)에 새로운 지원서류가 요구될 수 있음.
의료기기 등급 변경
Class IIB 군의 생체이식 시리즈 기기들은 Class III의 규정을 만족해야만 하며, 대부분의 소프트웨어는 Class I로 편입될 가능성이 낮음
위험물질 함유기기
발암 물질, 기형 발생 유발 물질, 생식장애 물질함유 기기에 대한 규정 강화
재사용 규제
사용하고 난 일회용품의 원료, 부품의 재사용에 대한 규제 강화
임상시험
임상연구 및 임상시험에 대한 새로운 규정이 마련될 예정이며, 임상 평가의 업데이트를 위해 중요한 시판 후 감시 데이터가 이용돼야 함
품질 관리
제조사는 기업 내에서 자격을 갖춘 품질관리사를 지정해 의료기기 제품에 대한 전문적인 지식을 보유하고 있어야 함.
인증기관 강화
유럽 차원에서 합의된 인증기관에 대한 새로운 시험 증명이 마련될 예정으로, 인증기관의 재검증이 이뤄지는바 인증기관 취소에 따른 인증취소 대비가 요구됨. 또한 고위험 의료기기 승인을 위한 특별 인증기관이 마련될 예정
□ CE 규정 개정 2: 체외 진단기(IVDR)
ㅇ IVDR 구조
- 유예기간 5년(2017~2021년)을 가지며 10장, 14부록으로 구성
* 10장(1. 사용분야 및 정의, 2. 제조업체, 유통업체 및 회원국에 대한 요구사항: 적합성평가 절차, 라벨 시판 후 임상추적, 사후 시장 감시, 3. 추적성, 기기 등록, 안전 및 임상 시험, EUDAMED 등록에 관한 사항, 4. 승인기관에 대한 요구사항, 5. 분류 및 적합성 평가, 6. 임상 평가 및 임상 시험, 7. 시장 감시 및 규제 리포팅, 8. “Medical Coordination Group“과 다른 전문가 집단, 회원국 간의 협력, 9. 기밀성, 데이터 보호, 처벌, 10. 최종 규정)
* 14부록(1. 일반 안전 및 성능 규제, 2. 기술 문서, 시판 후 감시에 대한 기술문서, 3. EU 선언의 적합성, 4. CE 마크의 적합성, 5. CE 마크의 적합성, 6. 인증기관의 요구사항, 7. 분류 기준, 8. 품질 관리 및 기술 서류 평가에 대한 적합성 평가, 9. 타입 검사에 따른 적합성 평가, 10. 상품 품질 보증에 따른 적합성 평가, 11. 인증기관에서 발급한 인증서, 12. 역량 평가 및 시판 후속 상황, 13. 중재적 임상실무 수행 연구 및 그 외 리스크 관련 연구, 14. 관련 테이블)
ㅇ 주요 변경 내용
주요 변경 사항
세부내용
의료기기 분류체계 변동
체외진단기기 분류에 따라 적합성 평가 절차가 달라짐. 기존 IVD 체계에서는 사용범위 따라 Class A와 Class B로 나뉘었다면, IVDR에서는 Class A(저 위험)~Class D(고 위험)으로 나뉨
적합성 평가 절차의 변동
저 위험군 기기(Class A)에 대한 적합성 평가 간소화로 인증기관을 거치지 않아도 됨. 단, Class A 이상의 기기 혹은 Class A 기기 중 살균의 조건이 필요한 기기(Class As)는 인증기관을 거쳐야 하며 이와 더불어 품질관리 시스템도 요구됨. 품질관리 시스템이 갖춰진 제조사는 Class D를 제외하고 제조사 스스로 적합성 평가 진행이 가능함
검증에 관한 변동
IVDR에서는 정확하고 추가적인 자료 작성이 요구되며 여기에는 과학적 데이터의 수집과 분석, 시판 후 후속 품질 관리에 대한 내용이 포함됨
고유식별코드(UDI) 도입
개정되는 의료기기규정과 마찬가지로 체외 진단기에도 적용될 예정
소프트웨어에 관한 규정
기존 IVD에서 규제에 포함되지 않았던 소프트웨어 관련 규정 강화
직원역량 강화
기업 내 자격을 갖춘 ‘특별 감사자‘ 지정
ㅇ 인터뷰 1: Emergo 의료기기 인허가 컨설팅
Q. 자사에 대해 소개 한다면?
A. Emergo는 전 세계 20개국에 지사를 두고 있는 글로벌 의료기기 인허가 컨설팅 그룹이다. Emergo는 의료기기 등록, 품질관리시스템 규제준수 및 감사, 인허가 컨설팅을 제공하는 기업이다.
Q. CE 개정에 대한 의견?
A. 이번 규정 강화 효과에 대해 매우 긍정적으로 평가한다. 발전하는 기술에 규제가 발맞춰가야 함은 물론, 의료기기는 인체에 미치는 영향이 매우 크므로 강력하게 규제해야 한다. 물론 기업 입장에서는 매우 골치 아픈 일이겠지만, 처음부터 품질관리·서류관리를 잘해오던 기업이라면 큰 실패 없이 진행할 수 있을 것이라 본다.
Q. CE 개정 시행 전이지만, 그에 대한 영향이 있는가?
A. 벌써부터 인증 신청·갱신이 지연되는 경우를 보는데,이를 통해 개정의 영향을 직접적으로 실감한다. 그와 더불어 개정관련 문의가 많이 들어오고 있다. 아직까지 다행히 인증 취소 등의 경우는 없으나 예의주시하고 있다.
Q. 한국 기업에 인증 변경에 관해 해주고 싶은 조언이 있다면?
A. CE 인증 준비 시 매우 중요한 점은 부록2의 기술 서류 부분 준비와(기기에 따라 필요하다면) 임상시험 준비를 철저히 해야 한다는 것이다. 처음부터 제대로 준비되지 않으면 다시 고치는 데 소요되는 시간과 에너지가 너무 크다. 이번 개정으로 기술문서* 부분이 더욱 세세하게 강화된바 그 중요성은 더 커질 것으로 본다. 또한 인증에서 중요한 것은 인증기관이다. 그러기 위해서는 인증기관과의 원활한 관계가 매우 중요하다. 인증 신청·갱신 시 외에도 자주 연락해 자사를 각인시기는 것이 좋다. 인증도 사람이 하는 일이다. 잘 알고 검증된 기업을 먼저 처리해 주고 싶지 않겠는가? 마지막으로 유예기간 3년은 전혀 길지 않은 시간이다. 지금부터 차근차근 꼼꼼히 준비하는 수밖에 없다.
Q. 기술문서에서 추가되는 부분은?
A. 아래와 같음.
- 기기 설명: 사진
- 식별: 고유식별코드(UDI)
- 적절한 사용 목적: 의도된 사용자, 환자 집단, 부속품, 사용금지 사유
- 물리적 원리: 새로운 기능에 대한 설명, 비슷한 제품에 대한 개요, 기존 제품에 대한 개요
- 구조: 필수 기능, 신체 접촉하는 제품일 경우 재료에 대한 설명, 기술 사양
- 라벨링: 카탈로그, 포장에 관한 내용
- 제조: 개발 및 생산에 관련된 모든 공급 및 하청업체 명시
- 위험관리: 위험관리 계획
- 소프트웨어: 다른 하드웨어와의 성능시험, 내부적 사용환경 시험ㅇ 인터뷰 2: 독일 내 한국 의료기기 업체
Q. 인증 및 등록절차는 어떻게 진행하고 있는가?
A. 본사는 규모가 있는 기업으로 한국 전담부서에서 맡아 진행하고 있다.
Q. CE 개정으로 인한 대비책은?
A. CE 규정 강화에 대한 소식은 인지하고 있었다. 시행을 앞둔 시점이지만 본사 담당 부서에서 대책을 준비한 것으로 안다. 인증이 만료되는 기기에 대해 원래 소요 기간보다 충분한 기간을 잡고 준비에 들어갔다.
Q. CE 개정 시행 전이지만, 그에 대한 영향이 있는가?
A. 앞서 언급한 대로 충분한 시간을 잡고 인증 갱신에 들어갔지만, 지난 11월 인증이 만료된 기기에 대해 아직까지도 인증이 나오지 않은 상태이다. 그로 인한 영업 애로사항과 손실이 막대하다. 현재 다른 심사기관에도 접촉해 인증 갱신을 신청 중이지만 올해 5~6월이 돼야 가능하다는 답변을 들었다. 현재로서는 기다리는 수밖에 없어 매우 답답한 심정이다.
Q. 다른 중소기업에 해주고 싶은 조언이 있다면?
A. 이미 시행됐다고 간주하고 준비해야 한다. 자사 기기에 적용되는 변경 사항을 꼼꼼히 챙기고, 긴 시간적 여유를 가지고 미리 미리 준비하기를 조언한다. 자사에 전담 부서가 없어 역량이 안 된다면 국내외 에이전시의 도움을 받는 것을 추천한다.□ 시사점
ㅇ 2015년 한국의 대독일 수출이 전년대비 14% 증가해 무역적자를 점점 줄이는 상황에서 독일 의료기기 시장은 2017년에도 4%대의 꾸준한 성장이 예상되고 있어 향후 한국 기업의 수출 증가가 예상됨.- 특히, 자기공명 촬영기, 치과용품, 컴퓨터 단층촬영기 등의 꾸준한 수출 증가가 예상됨.
ㅇ 독일 시장 및 유럽 시장에 진출하려고 하는 국내 기업은 이번 'EU 의료기기, 체외진단 의료기기 CE 규정' 변동사항을 면밀히 검토해 절저한 준비를 해야할 것임.- 특히 변경된 등급확인, 의료기기 고유식별코드(UDI) 적용준비, 새로운 라벨링 적용, 임상실험자료 준비, 유럽 데이터베이스 등록 준비에 각별한 주의가 요구됨.
- 현재 모든 인증기관에 대한 MDR, IVDR의 개정 요구사항을 기준으로 재평가가 이뤄지는바 인증기관의 인증 취소를 대비한 대처가 필요
ㅇ 준비에 만전을 기하고 충분한 시간적 여유를 가지고 진행하는 것이 현명함.
- 한국 기업 사례로 보았듯 인증 갱신임에도 불구하고 소요되는 시간이 증가함. 인증으로 인한 영업손실을 막기 위해 보통 소요 시간보다 2~3배 걸릴 것을 예상하고 진행할 것을 추천
인증 신청 시 일반적 소요 시간구분
Class I
(비살균, 비계측)
Class I
(살균, 계측)
Class ll A
Class ll B
Class lll
서류 제출 후 소요기간
1개월 이내
3~5개월
3~5개월
3~6개월
6~9개월
유효기간
없음
3년
3년
3년
3년
인증 갱신 신청
없음
최소 6개월 전
최소 6개월 전
최소 6개월 전
최소 6개월 전
자료원: 독일 의료기술 협회, 글로벌 트레이드 아틀라스, Johner Institut, 이머고, 인터뷰 및 KOTRa 함부르크 무역관 자료 종합
<저작권자 : ⓒ KOTRA & KOTRA 해외시장뉴스>

KOTRA의 저작물인 (新 EU 의료기기 규정, 하반기 전면 시행에 미리 대비해야 )의 경우 ‘공공누리 제4 유형: 출처표시+상업적 이용금지+변경금지’ 조건에 따라 이용할 수 있습니다. 다만, 사진, 이미지의 경우 제3자에게 저작권이 있으므로 사용할 수 없습니다.
-
1
초음파 영상진단기
프랑스 2020-07-10
-
2
스위스-EU 의료기기 규정 개정, 상세 의미와 대응방안
스위스 2021-08-04
-
3
체외진단 의료기기 규제 강화하는 유럽
네덜란드 2018-06-15
-
4
이탈리아 미용기기 제품 시장동향
이탈리아 2020-10-07
-
5
이집트 의료기기 등록 규정 및 절차 안내
이집트 2016-12-07
-
6
독일, 의료 IT 시장이 열린다
독일 2017-03-27
-
1
2025년 독일 철강산업 정보
독일 2025-04-01
-
2
2024년 독일 바이오헬스 산업 정보
독일 2024-12-20
-
3
2024년 독일 반도체 산업 정보
독일 2024-12-11
-
4
2024년 독일 IT 산업 정보
독일 2024-08-14
-
5
2024 독일 항공 우주 산업 정보
독일 2024-07-14
-
6
2021 독일 수소산업 정보
독일 2021-12-31